
Documentation on Moderna’s non-clinical summaries and biodistribution studies from Module 2 of Spikevax BLA has been made available via FOIA by Judicial Watch.

Judicial Watch announced on December 13, 2022 that it received 699 pages of records from the Department of Health and Human Services (HHS) regarding data Moderna submitted to the Food and Drug Administration (FDA) on its mRNA COVID-19 vaccine. The documents also reveal Moderna elected not to conduct a number of standard pharmacological studies on the laboratory test animals. The records include a “Nonclinical Overview” prepared by Moderna and submitted to the FDA for approval of its vaccine revealing that a number of rats were born with skeletal deformations, known as “wavy ribs” and “rib nodules,” to mothers injected with the mRNA vaccine. The study dismissed the anomalies as “not considered adverse”, without any additional data demonstrating exclusion of this risk. The FDA published a direct lie in the regulatory summary statement on Spikevax.
Background: Relevant FDA Guidance for Development of Gene Therapies and Covid-19 Vaccines.
Non-clinical testing is part of the drug or vaccine development process during which the product is tested in cell lines and animals. The FDA publishes Industry Guidance documents describing regulatory knowledge and expectations for pharmaceutical manufacturers developing certain classes of products. Due to the innovative nature of the pharmaceutical research and development, Guidance documents are generally non-binding recommendations and the precise scope of each phase of development is negotiated by the pharmaceutical sponsor with the regulators. This process is supposed to consider available scientific, regulatory, and clinical (if other products in the class have been approved) knowledge about the product class, and it’s potential and known risks. Parts of the FDA Guidance are enforceable.
The FDA has been publishing guidance documents for cellular and gene therapies starting in 1998. Therefore, an extensive body of regulatory knowledge regarding this product class has been available for the past 20+ years, the most recent pre-covid pandemic ones being 2013 (Preclinical Guidance) and 2015 (Clinical Guidance). Note that many other rules and regulations apply to the development, manufacture, labeling and testing of biological products containing genetic materials and are expected to be considered by a BLA applicant for products containing genetic materials. This report addresses only preclinical development issues.
The 2015 “Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products” lists potential of these products to cause serious safety issues, including death, potential to promote cancer, uncontrollable and irreversible expression of proteins, genotoxicity, reproductive harm, and potential for transmission to other individuals through “shedding,” among many others. Both the manufacturers and regulators are expected to anticipate these risks and design preclinical testing programs to exclude or fully characterize the harms to humans before the product can be even administered in controlled setting of clinical trials, and definitely prior to authorization and mass deployment.
Covid-19 vaccines utilize the same chemical methods, mechanism of action and manufacturing “platform” technology and its implementation as do gene therapies. In fact, Moderna’s products were classified as gene therapies prior to and even in 2020 per company’s own shareholder disclosures.
Regarding covid mRNA/DNA vaccines specifically, the FDA published “Development and Licensure of Vaccines to Prevent Covid-19” in June 2020. This guidance document is final, was implemented without prior public participation, and is intended to remain in effect for the duration of the public health emergency related to COVID-19 declared by the Secretary of Health and Human Services (HHS) on January 31, 2020, effective January 27, 2020.
Regarding the nonclinical assessments for Covid-19 Vaccines (2020) the FDA guidance states that:
· (P.6)“For a COVID-19 vaccine candidate consisting of a novel product type and for which no prior nonclinical and clinical data are available, nonclinical safety studies will be required prior to proceeding to FIH [first-in-human] clinical trials 21 CFR 312.23(a)(8).
· (P.7)“When needed to support proceeding to FIH clinical trials, nonclinical safety assessments including toxicity and local tolerance studies must be conducted under conditions consistent with regulations prescribing good laboratory practices for conducting nonclinical (sic) laboratory studies (GLP) (21 CFR Part 58). Such studies should be completed and analysed (sic) prior to initiation of FIH clinical trials. When toxicology studies do not adequately characterize risk, additional safety testing should be conducted as appropriate.”
· (P.7) “Use of COVID-19 preventive vaccines in pregnancy and in women of childbearing potential will be an important consideration for vaccination programs. Therefore, FDA recommends that prior to enrolling pregnant women and women of childbearing potential who are not actively avoiding pregnancy in clinical trials, sponsors conduct developmental and reproductive toxicity (DART) studies with their respective COVID-19 vaccine candidate. Alternatively, sponsors may submit available data from DART studies with a similar product using comparable platform technology if, after consultation with the agency, the agency agrees those data are scientifically sufficient.”
· (P.8) “Current knowledge and understanding of the potential risk of COVID-19 vaccine associated ERD [Enhanced Respiratory Disease] is limited, as is understanding of the value of available animal models in predicting the likelihood of such occurrence in humans. Nevertheless, studies in animal models (e.g., rodents and non-human primates) are considered important to address the potential for vaccine-associated ERD.”
The agency also stated that where the guidance says “should” – those sentences describe nonbinding recommendations, while where it says “must” and cites the applicable law, it describes enforceable items.
Review of Moderna’s FOIA Production of Nonclinical Summaries vs Existing FDA Guidance:
1. Spikevax mRNA-1273 is a novel pharmaceutical chemical entity, and the requirement for completion of nonclinical safety studies prior to first-in-human trials was not met.
There were no nonclinical or clinical data available for mRNA-1273 prior to 2020, and no other similar product was approved anywhere in the world. The only previously approved product containing RNA is Onpattro (www.onpattro.com). It contains small, non-coding RNAs called short interfering or siRNAs, typically 20-24 nucleotides in length. It is indicated for polyneuropathy due to an extremely rare, hereditary, and fatal “orphan disease” amyloidosis that is estimated to affect just 50,000 people worldwide. The mechanism of siRNA is different than that of modified or mRNA, which is a very large molecule (~3000-4000 nucleotides). As opposed to mRNA, siRNAs do not code for proteins, they “silence” or prevent translation of target genes.
Moderna’s mRNA-1273 formulated in lipid nanoparticle is an entirely novel chemical entity. While the regulatory opinion on this topic has not yet been disclosed for Moderna, the European Medicines Agency reviewers wrote the following opinion regarding Pfizer’s mRNA-LNP product’s novelty (see EMA document in Attachment):
“The modified mRNA in the COVID-19 mRNA Vaccine is a chemical active substance that has not been previously authorized in medicinal products in the European Union. From a chemical structure point of view, the modified mRNA is not related to any other authorized substances. It is not structurally related as a salt, ester, ether, isomer, mixture of isomers, complex, or derivative of an already approved active substance in the European Union.
The modified mRNA is not an active metabolite of any active substance(s) approved in the European Union. The modified mRNA is not a pro-drug for any existing agent. The administration of the applied active substance does not expose patients to the same therapeutic moiety as already authorized active substance(s) in the European Union.
A justification for these claims is provided in accordance with the “Reflection paper on the chemical structure and properties criteria to be considered for the evaluation of new active substance (NAS) status of chemical substances” (EMA/CHMP/QWP/104223/2015), COVID-19 mRNA Vaccine is therefore classified as a New Active Substance and considered to be new in itself.” (EMA Assessment report on the claim of new active substance (NAS) status of 5’capped mRNA encoding full-length SRAS-CoV-2 Spike protein contained in COVID-19 mRNA Vaccine BioNTech, Nov 11, 2020.)
Moderna’s Spikevax is the same class of chemical entity and product, utilizing same design, same target antigen, similar lipid nanoparticle mechanism of delivery, and claims an identical mechanism of action.
Therefore, based on the FDA Guidance, nonclinical safety studies were required for Moderna mRNA-1273 before initiation of “first-in-human” (FIH) clinical trials. This requirement is enforceable as the applicable law is cited in the guidance (21 CFR312.23(a)(8).
According to the Moderna HHS production, as well as FDA’s “January 30, 2022, Summary Basis for Regulatory Action – Spikevax”, the Investigational New Drug Application (CFR 21 Part 312) to conduct Phase 1 human trials was opened on February 20, 2020 (IND# 19635). It was opened on behalf of Moderna by the National Institutes of Health (DMID). The second IND for Spikevax was opened on April 27, 2020, for Phase 2 human trials (IND# 19745). It is not clear why two IND numbers exist for a single vaccine product and why US Government owns one of them.
Based on this timeline, the FIH studies for Moderna were approved to proceed and initiated on February 20, 2020. According to the CDC, as of January 11, 2020, Chinese health authorities said they’ve identified more than 40 human infections as part of this outbreak that was first reported on December 31. The World Health Organization announced the preliminary identification of the novel coronavirus on January 9. The record of Wuhan-Hu-1 includes sequence data, annotation, and metadata from this virus isolated from a patient approximately two weeks prior. At the time, no animal studies were completed for mRNA-1273 and this non-clinical part of Moderna’s dossier could not have been reviewed properly by the FDA to inform the IND decision.
Therefore, the enforceable requirement for a scientifically rigorous dossier of nonclinical studies to be completed prior to FIH for a novel product was not met by Moderna.
2. The only GLP toxicology study conducted with mRNA-1273 was the reproductive toxicity study, in female animals only. No other mRNA-1273 nonclinical studies were performed under GLP.
The package of materials obtained from HHS contained 699 pages of nonclinical studies and test results that were used by the FDA to clear Moderna’s mRNA platform-based mRNA-1273 (Moderna’s Covid-19 vaccine, or SPIKEVAX).
The production contained only one complete study report: “A Single Dose Intramuscular Injection Tissue Distribution Study of mRNA-1647 in Male Sprague-Dawley Rats”, a non-Good Laboratory Practice (non-GLP) compliant study conducted in 2017 at Charles River Laboratories Montreal ULC, Sherbrooke Site (CR SHB) 1580 Ida-Metivier Sherbrooke, QC J1E 0B5 Canada. The remainder of the package contained narrative summaries of other non-clinical studies included in the BLA submission.
Studies included in the package contained numerous other experimental and unapproved mRNA products unrelated to Sars-Cov-2 or covid illness.
There were three separate versions of Module 2.4 (standard part of BLA) included, and many sections appear to be missing. It is not clear why multiple versions are included, and there is no explanation provided as to which version specifically was used for the approval of SPIKEVAX by the FDA. Module 2.4 starts on p. FDA-CBER-2021-4379-0001130 and continues to p.-0001160, the second and third copies start on 0001462 and up to -0001548. However, all three versions contain similar text summaries.
Out of approximately 25 studies included in nonclinical program (See Exhibit 1 at the end of this document), there was a single, non-GLP biodistribution study in male rats, conducted at the Charles River facility in Canada. The study was for an irrelevant drug substance test article, mRNA-1674 and not Spikevax mRNA-1273, formulated in the lipid nanoparticle (LNP) claimed to be substantially similar to the Spikevax LNP formulation. This experimental and unapproved product is a construct of 6 different experimental mRNAs studied for vaccination against cytomegalovirus in 2017 and never approved for market anywhere in the world. This study demonstrated distribution of the lipid nanoparticles throughout the entire body and accumulation in all major organ systems, except the kidney. This indicates, that if LNP is carrying the Spikevax mRNA-1273 it will likewise be distributed throughout the body and accumulate in the same organs.
Notably, the study assessed only male animals and not females, therefore no information about biodistribution in females was made available to the FDA, and specifically, no information about exposure differences or accumulation in female reproductive organs was ever obtained. The reproductive toxicology study, discussed below (part 3), while conducted in only females, did not make biodistribution measurements.
In the biodistribution study, high LNP concentrations were observed in lymph nodes and spleen and persisted in those organs at 3 days after the injection. The study was stopped before full clearance could be observed, therefore, no knowledge exists on the full-time course of the biodistribution. Other organs where vaccine product was detected included bone marrow, brain, eye, heart, small intestine, liver, lung, stomach, and testes. Given that the LNPs and mRNA-1647 were detected in all these issues, it is reasonable to assume that the LNPs carrying mRNA-1273 likewise would distribute in the same way, and therefore, the spike protein would be expressed by the cells in those critical organ systems with unpredictable and possibly catastrophic effects.
It appears that no additional biodistribution studies were conducted with the final implementation of mRNA-1273, and therefore it is unknown what proteins are expressed in the tissues and organs. Therefore, the mechanism of action of the product has not been fully demonstrated.
There was a single, non-GLP repeat dose toxicity study with mRNA-1273, however the study was not completed at the time of the FOIA production (April 2022). This study is titled “5 weeks (2 doses) repeat immunogenicity and toxicity study” and was conducted in rats. This study is described very briefly and refers to full reports in Module 2.6.2, which was not included in the HHS production. The summary of the results includes the following statement on safety observations: “There were no mRNA-1273-related effects on body weight. mRNA-1273-related clinical signs were observed on Day 1 and Day 22, starting at the 30 µg/dose. Clinical signs consisting of transient dose-dependent injection site edema with or without hindlimb impairment were observed at approximately 24 hours post-dose and generally resolved within 7 days after dose administration. mRNA-1273-related clinical pathology changes associated with inflammation, including increased neutrophils, eosinophils, and/or globulin, were observed starting at the 30 µg/dose. Other mild mRNA-1273-related changes observed at 30, 60, and/or 100 µg/dose consisted of decreased red cell mass, reticulocytes, and lymphocytes and increased creatinine, triglyceride, cholesterol, and/or glucose”.
Found in a separate part of the data package, an updated paragraph on this study results reads that:
“Post-mortem test article-related and generally dose-dependent changes in organ weights and macroscopic and microscopic findings were observed at ≥ 8.9 µg/dose. Organ weight increases were observed in the spleen, liver, and adrenal gland. Organ weight changes were generally reversing by the end of the 2-week recovery period. Macroscopic changes included skin thickening at the injection site and enlarged lymph nodes. Injection site changes completely recovered, and lymph node changes were recovering by the end of the 2-week recovery period. Microscopic changes included mixed cell inflammation at the injection site; increased cellularity and mixed cell inflammation in the inguinal, iliac, and popliteal lymph nodes; decreased cellularity in the splenic periarteriolar lymphoid sheath; increased myeloid cellularity in the bone marrow; and hepatocyte vacuolation and Kupffer cell hypertrophy in the liver. Microscopic changes were generally reversing by the end of the 2-week recovery period.”
These markers are indicative of possible tissue damage, systemic inflammation, and potential severe safety issues, and they are also dose dependent. Statements like “lymph node changes were recovering” are an admission that the product is associated with a potentially serious safety signal which was still ongoing when the study was terminated. There were no follow up studies to assess the risks and exclude toxicity to these organ classes.
There was a single GLP reproductive toxicity using mRNA-1273, however in the study only female animals were vaccinated. Males were not (discussed in part 3 below).
The remainder of the package contained 10 primary pharmacology studies with mRNA-1273, however all were non-GLP (see Exhibit 1, Table 1). These studies are discussed below. The remainder of the production contained 6 GLP repeat dose toxicity studies that used irrelevant test article – other experimental and previously unapproved mRNAs (See Exhibit 1, Table 3), 4 in-vitro genotoxicity experiments that included only 2 components of the lipid nanoparticle formulation (SM-102 and PEG2000), but no other parts of the final product in its final formulation, and 2 in-vivo genotoxicity studies with irrelevant test articles.
No carcinogenicity studies with final product formulation were conducted. There were no safety pharmacology assessments for any organ classes such as cardiovascular, CNS, liver, spleen, etc.
Therefore, the enforceable requirement for GLP studies with the correct test article in final commercially manufactured formulation has not been met in 24 out of the 25 studies. Furthermore, the studies have not excluded toxicity risks to major organ systems, and no additional studies to evaluate and exclude those risks were conducted prior to large scale administration of the product in human studies.
3. The reproductive toxicity study of mRNA-1273, in female animals only, demonstrated that risks to pregnancy, and a statistically significant increase in fetal developmental abnormalities.
Moderna conducted Reproductive Toxicology study in pregnant and lactating rats using human dose of 100 mcg mRNA-1273 (described on p. -0001150 of the HHS production).
In this study, the reproductive toxicities were assessed for females only, males were not vaccinated. Therefore, no assessment of male reproductive toxicities have been made at all.
The full study report was not included in the production. In the narrative summary of the findings, Moderna wrote:
“High IgG antibodies to SARS-CoV-2 S-2P were also observed in GD 21 F1 fetuses and LD 21 F1 pups, indicating strong transfer of antibodies from dam to fetus and from dam to pup.”
Safety assessments in the study appear to be very limited, however, the following findings are described by Moderna: The mothers lost fur and appetite after vaccine administration, and it persisted for several days. There is no information on when it was fully resolved since the study was terminated before this could be assessed.
In the rat pups, the following skeletal malformations were observed:
“In the F1 generation [rat pups], there were no mRNA-1273-related effects or changes in the following parameters: mortality, body weight, clinical observations, macroscopic observations, gross pathology, external or visceral malformations or variations, skeletal malformations, and mean number of ossification sites per fetus per litter. mRNA-1273-related variations in skeletal examination included statistically significant increases in the number of F1 rats with 1 or more wavy ribs and 1 or more rib nodules. Wavy ribs appeared in 6 fetuses and 4 litters with a fetal prevalence of 4.03% and a litter prevalence of 18.2%. Rib nodules appeared in 5 of those 6 fetuses.”

The skeletal malformations were temporally associated to the days when highest toxicity was observed in the mothers: “Maternal toxicity in the form of clinical observations was observed for 5 days following the last dose (GD 13), correlating with the most sensitive period for rib development in rats (GDs 14 to 17)”.
The FDA included the following statement in the Basis for Regulatory Action Summary document (Section 4: Non-clinical Reproductive Toxicology, p.14):
“No vaccine-related fetal malformations or variations and no adverse effect on postnatal development were observed in the study. Immunoglobulin G (IgG) responses to the pre-fusion stabilized spike protein antigen following immunization were observed in maternal samples and F1 generation rats indicating transfer of antibodies from mother to fetus and from mother to nursing pups.”
The basis for agency’s dismissal of the risk of skeletal malformations in the vaccinated rat offspring is not clear, given Moderna’s admission of statistical significance of this signal, as well as temporal association with the highest levels of antigen expression in the mothers.
In summary, the vaccine-derived antibodies transfer from mother to child. It was never assessed by Moderna whether the LNPs, mRNA, and spike proteins transfer as well, but it is reasonable to assume that they do due to the claimed mechanism of action of these products. This should have been tested, and risks to the child should have been assessed, as the baby’s major metabolizing organs, such as the liver, are very small. Dose finding studies for Moderna are still ongoing and incomplete. An overdose through transfer and high expression of spike protein could pose serious life-threatening risks to a baby. This was never disclosed to the pregnant and breastfeeding women who were being coerced into receiving these injections.
4. The vaccine-induced antibody-enhanced disease was identified as a serious risk by the FDA. This risk was not excluded by Moderna nonclinical studies due to the absence of positive control and unvalidated analytical methods used.
Prior to 2020, Moderna had not been able to bring any products to market, and few of its development candidates ever progressed to late phase clinical trials in humans. None have reached the regulatory approval phase. Its entire product development history was marked by numerous failures despite approximately $5 billion dollars and 10+ years spent in research.
Notably, its mRNA-based vaccines were associated with the antibody-enhancement (ADE) phenomenon. ADE and ERD both describe the same clinical concern – worsening of the disease severity after vaccination for the disease. One such example includes Moderna’s preclinical study of mRNA-based zika vaccine in which vaccinated mice all “uniformly [suffered from] lethal infection and severe disease due to antibody enhancement.” The scientists were able to develop a vaccine type (IgEsig-prM-E FL) that generated protection against Zika and “resulted in significantly less morbidity and mortality,” although all versions of the vaccine unequivocally led to some level of ADE. By day 5, survival rate for mice vaccinated with IgEsig-prM-E FL dropped to 80%, meaning 20% of the vaccinated mice had died due to ADE induced by the vaccine.
The Primary Pharmacology section in the HHS production of Moderna’s nonclinical studies (2.4.2 Pharmacology, Table 1) included 10 studies evaluating immunogenicity, protection from viral replication (declared mechanism of action), and to evaluate potential for vaccine-associated enhanced respiratory disease. These studies included the correct test article (mRNA-1273), however, all were non-GLP compliant. The results of these studies are briefly summarized in the text of the document package; however, the study reports are not provided.
On p.-000150, Moderna stated that “there were no established animal models” for SARS-Cov-2 virus due to its extreme novelty. In the next sentence, it appears that despite the extreme novelty of the virus, Dr. Ralph Baric at the University of North Carolina at Chapel Hill possessed an already mouse-adapted SARS-Cov-2 virus strain and provided it for some of the Moderna’s experiments.
Elsewhere, on p.-0001468, Moderna stated that the EDR concern was triggered by preclinical work on SARS-CoV and MERS-CoV vaccines, among others – so it seems the company is contradicting its own statements about lack of preclinical models of SARS. While discussing the ERD risk, Moderna stated that their own study results were unreliable due to the invalidity of the assays and absence of the positive control in their experiments: “As SARS-CoV-2 neutralization assays are, to this point, still highly variable and in the process of being further developed, optimized and validated, study measurements should not be considered a strong predictor of clinical outcomes, especially in the absence of results from a positive control that has demonstrated disease enhancement”.
Therefore, the risk of the ERD was not excluded, and not characterized in these studies.
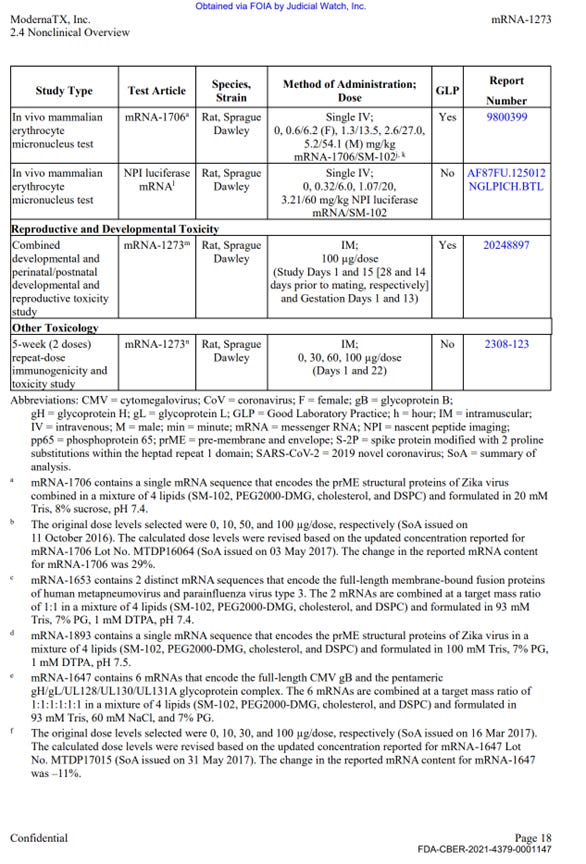
EXHIBIT 1.
Tables summarizing the scope of the program from HHS Production of Moderna’s Nonclinical Studies.
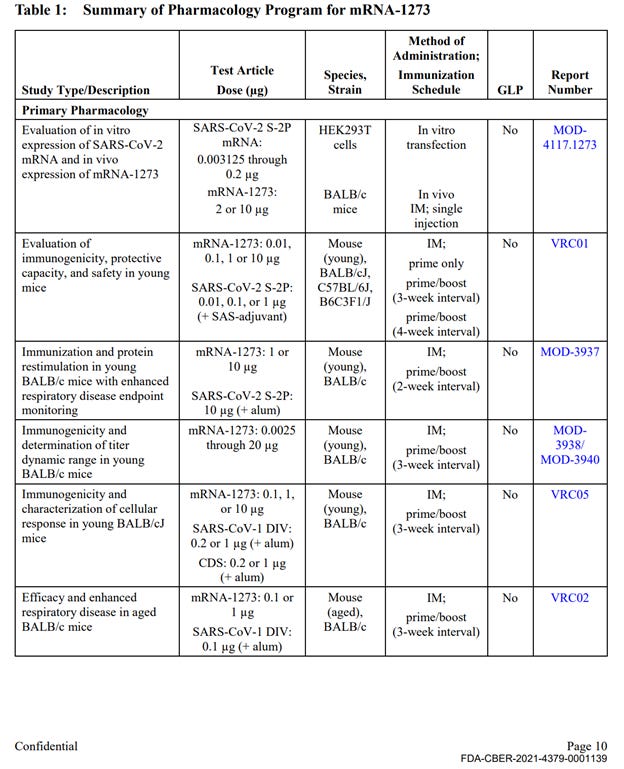
Table 1 continued:
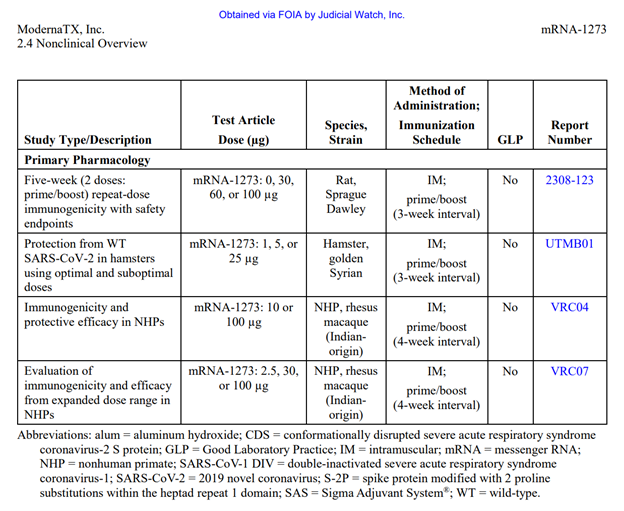
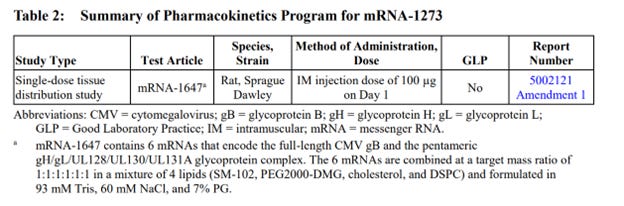
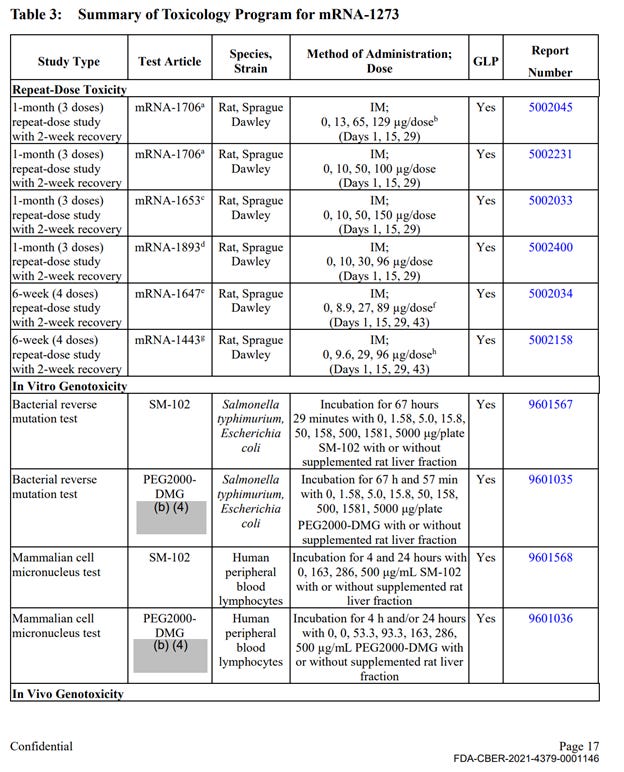
Table 3 continued:
Source – https://sashalatypova.substack.com/p/my-affidavit-on-modernas-nonclinical