
Patients and doctors expect drug regulators to provide an unbiased, rigorous assessment of investigational medicines before they hit the market. But do they have sufficient independence from the companies they are meant to regulate? Maryanne Demasiinvestigates
Over the past decades, regulatory agencies have seen large proportions of their budgets funded by the industry they are sworn to regulate.
In 1992, the US Congress passed the Prescription Drug User Fee Act (PDUFA), allowing industry to fund the US Food and Drug Administration (FDA) directly through “user fees” intended to support the cost of swiftly reviewing drug applications. With the act, the FDA moved from a fully taxpayer funded entity to one supplemented by industry money. Net PDUFA fees collected have increased 30 fold—from around $29m in 1993 to $884m in 2016.1
In Europe, industry fees funded 20% of the new EU-wide regulator, the European Medicines Agency (EMA), in 1995. By 2010 that had risen to 75%; today it is 89%.2
In 2005 in the UK, the House of Commons’ health committee evaluated the influence of the drug industry on health policy, including the Medicines and Healthcare Products Regulatory Agency (MHRA).3 The committee was concerned that industry funding could lead the agency to “lose sight of the need to protect and promote public health above all else as it seeks to win fee income from the companies.” But nearly two decades on, little has changed, and industry funding of drug regulators has become the international norm.
The BMJ asked six leading regulators, in Australia, Canada, Europe, Japan, the UK, and US, a series of questions about their funding, transparency in their decision making (and of data), and the rate at which new drugs are approved. We found that industry money permeates the globe’s leading regulators, raising questions about their independence, especially in the wake of a string of drug and device scandals.
Industry fees
Industry money saturates the globe’s leading regulators. The BMJ found that the majority of regulators’ budget—particularly the portion focused on drugs—is derived from industry fees (table 1).
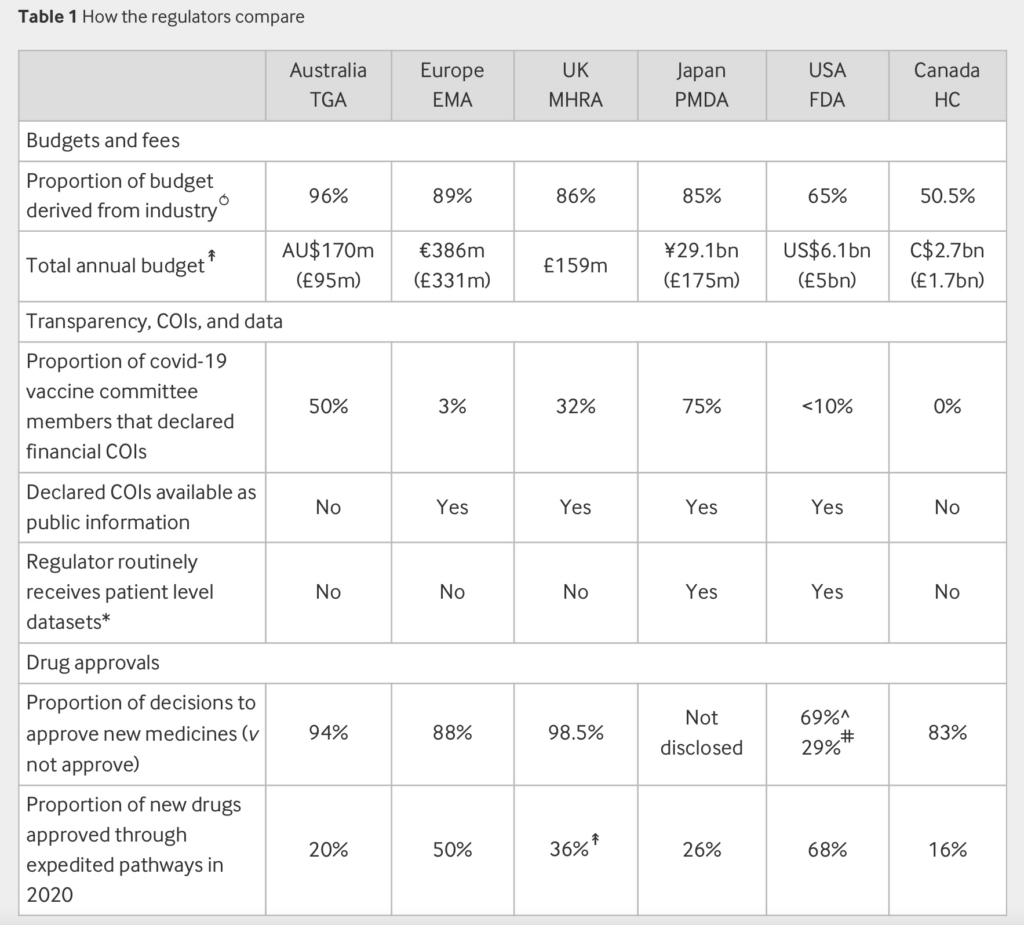
Note: Data sources and methods are detailed in the supplemental file
↟Data refer to the year 2021 calendar year or 2020-2021 fiscal year
⥀Many agencies regulate beyond medical products (for example, food); where possible (US, Canada), we used the proportion of the human drugs budget
FDA: US Food and Drug Administration; EMA: European Medicines Agency; TGA: Therapeutic Goods Administration; HC: Health Canada; MHRA: Medicines and Healthcare Products Regulatory Agency; PMDA: Pharmaceuticals and Medical Devices Agency
Of the six regulators, Australia had the highest proportion of budget from industry fees (96%) and in 2020-2021 approved more than nine of every 10 drug company applications. Australia’s Therapeutic Goods Administration (TGA) firmly denies that its almost exclusive reliance on pharmaceutical industry funding is a conflict of interest (COI). In response to a query, the agency said, “All fees and charges are prescribed in our legislation. To provide transparency, the TGA fees and charges are published on the TGA website.”
But for decades academics have raised questions about the influence funding has on regulatory decisions, especially in the wake of a string of drug and device scandals—including opioids, Alzheimer’s drugs, influenza antivirals, pelvic mesh, joint prostheses, breast and contraceptive implants, cardiac stents, and pacemakers.4567An analysis of three decades of PDUFA in the US has shown how a reliance on industry fees is contributing to a decline in evidentiary standards, ultimately harming patients.8 In Australia, experts have called for a complete overhaul of the TGA’s structure and function, arguing that the agency has become too close to industry.
Sociologist Donald Light of Rowan University in New Jersey, US, who has spent decades studying drug regulation, says, “Like the FDA, the TGA was founded to be an independent institute. However, being largely funded by fees from the companies whose products it is charged to evaluate is a fundamental conflict of interest and a prime example of institutional corruption.”
Light says the problem with drug regulators is widespread. Even the FDA—the most well funded regulator—reports 65% of its funding for the evaluation of drugs comes from industry user fees (table 1),9 and over the years user fees have expanded to generic drugs, biosimilars, and medical devices.
“It’s the opposite of having a trustworthy organisation independently and rigorously assessing medicines. They’re not rigorous, they’re not independent, they are selective, and they withhold data. Doctors and patients must appreciate how deeply and extensively drug regulators can’t be trusted so long as they are captured by industry funding.”
External advisers
Concern over COIs is not just directed at those who work for the regulators but extends to the advisory panels intended to provide regulators with independent expert advice.1011 A BMJ investigation last year found several expert advisers for covid-19 vaccine advisory committees in the UK and US had financial ties with vaccine manufacturers—ties the regulators judged as acceptable.11 A large study that investigated the impact of COIs among FDA advisory committee members over 15 years found that those with financial interests solely in the sponsoring firm were more likely to vote in favour of the sponsor’s product,12 and that people who served on advisory boards solely for the sponsor were significantly more likely to vote in favour of the sponsor’s product. Research exploring the matter from a cross-national comparative perspective is lacking, however.
In Australia, the membership of the TGA’s Advisory Committee on Vaccines is published on the agency’s website. The forms for recording past and current financial and non-financial interests are not, however, made public. A Freedom of Information (FOI) Act request for their financial disclosures in August 2020 had names and details of the disclosures redacted.13 After seeking additional details, the TGA indicated that this was “personal information” and therefore usually exempt under the FOI act. Subsequently, panel members were approached directly by email and asked whether they would be willing to publish their declarations, but there was no response. Instead, they referred the enquiry back to the TGA which was willing to reveal that 5 of 10 committee members disclosed COIs—but did not say which members or provide any specifics, adding that “these interests usually do not give rise to a conflict.” The agency’s policy allows for excluding members from certain meetings because of a COI, but details of the COI and reasons for the exclusion are not published.
Joel Lexchin, a drug policy researcher at York University in Toronto, says, “People should know about any financial COIs that those giving advice have so that they can evaluate whether those COIs have influenced the advice they are hearing. People need to be able to trust what they hear from public health officials and a lack of transparency erodes trust.”
Of the six major regulators approached by The BMJ, only Canada’s drug regulators did not routinely seek advice from an independent committee and its evaluation team was the only one completely free of financial COIs. European, Japanese, and UK regulators publish a list of members with their full declarations online for public access, while the FDA judges COIs on a meeting-by-meeting basis and can grant waivers allowing participation of members (table 1).
Transparency, conflicts of interest, and data
Over the past decade, there have been improvements in the transparency and accessibility of trial data. Today the EMA and Health Canada (HC) both post to their website substantial amounts of clinical data received by the drug sponsor.1415 In addition, Japan’s Pharmaceuticals and Medical Devices Agency (PMDA) posts non-clinical data summaries.16
Most regulatory agencies do not, however, undertake their own assessment of individual patient data, but rather rely on summaries prepared by the drug sponsor. The TGA, for example, says it conducts its covid-19 vaccine assessments based on “the information provided by the vaccine’s sponsor.” According to a FOI request from last May, the TGA said it had not seen the source data from the covid-19 vaccine trials. Rather, the agency evaluated the manufacturer’s “aggregate or pooled data.” The TGA does not have the individual participant level datasets pertaining to the covid-19 vaccine trials,17 which are held by the vaccine manufacturer.18
“The TGA should not be relying on the analysis of that data produced by the drug companies. Rather the TGA should be reanalysing the source data,” says Lexchin. “Further, the TGA should be holding public hearings before new drugs are approved so that it can hear from members of the public and outside scientists.”
The TGA is hardly alone. Among global regulators, only two—the FDA and PMDA—routinely obtain patient level datasets. And neither proactively publish these data. Recently, a group of more than 80 professors and researchers called the Public Health and Medical Professionals for Transparency sued the FDA for access to all the data which the agency used to grant licensure for Pfizer’s covid-19 vaccine.19 The FDA argued that the burden on the agency was too great and requested that it be allowed to release appropriately redacted documents at the rate of 500 pages a month, a speed that would take approximately 75 years to complete. In a win for transparency advocates, this was overturned by a US Federal Court Judge, ruling that the FDA would need to turn over all the appropriately redacted data within eight months. Pfizer sought to intervene to ensure “information that is exempt from disclosure under the FOI act is not disclosed inappropriately,” but its request was denied.
Speedy approvals
Following the AIDS crisis of the 1980s and 1990s, PDUFA “user fees” were introduced in the US to fund additional staff to help speed the approval of new treatments. Since then, there has been concern over the way it moulded the regulatory review process—for example, by creating “PDUFA dates,” deadlines for the FDA to review applications, and a host of “expedited pathways” for speeding drugs to market. The practice is now a global norm.
Today, all major regulators offer expedited pathways that are used in a significant proportion of new drug approvals. In 2020, 68% of drug approvals in the US were through expedited pathways, 50% in Europe, and 36% in the UK.
Accelerated approval processes have resulted in new drugs that were more likely to be withdrawn for safety reasons, more likely to carry a subsequent black box warning, and more likely to have one or more dosage forms voluntarily discontinued by the manufacturer.202122
“One reason why drugs approved by the FDA so close to the deadline may have had more safety problems is that the FDA reviewers were afraid of going over the deadline for making a decision and thereby jeopardising the revenue that the FDA gets from drug companies,” says Lexchin.
Aaron Kesselheim, professor of medicine at Brigham and Women’s Hospital and Harvard Medical School, adds that accelerated approvals generally have a lower burden of proof for efficacy.
“The accelerated approval pathway explicitly changes the underlying efficacy ‘standard’ in that it allows approval based on changes to a surrogate measure that is not well validated, and is only reasonably likely to predict clinical benefit,” says Kesselheim who resigned from an FDA advisory committee last year in protest over the agency’s approval of a controversial Alzheimer’s drug. Following the committee’s vote against approval, the FDA shifted the goal posts, approving aducanumab through an accelerated approval based on the disputed surrogate measure of lowered visible β-amyloid protein levels.23
Courtney Davis, a medical and political sociologist at the Kings College London, says that a general taxation or a drug company levy would be better options to fund regulators. “PDUFA is the worst kind of arrangement since it allows industry to shape FDA policies and priorities in a very direct way. Each time PDUFA was reauthorised, industry had a seat at the table to renegotiate the terms of its funding and determine which performance metrics and goals the agency should be evaluated by. Hence the FDA’s focus on making quicker and quicker approval decisions—even for drugs not judged to be therapeutically important for patients.”
The regulator-industry revolving door
Critics argue that regulatory capture is not only being baked in by the way in which agencies are funded, but also staffed. A “revolving door” has seen many agency officials end up working or consulting for the same companies they regulated.
At the FDA, generally regarded as the world’s premier regulator, nine out of 10 of its past commissioners between 2006 and 2019 went on to secure roles linked with pharmaceutical companies,24 and its 11th and most recent, Stephen Hahn, is working for Flagship Pioneering, a company that acts as an incubator for new biopharmaceutical companies.
In February, the US Senate narrowly confirmed Robert Califf, a cardiologist, to lead the FDA, a position he previously held under the Obama administration. Califf’s rehiring led some senators to argue that his ties to the pharmaceutical industry made him unfit for the role. Financial disclosure forms show Califf was paid $2.7m by Verily Life Sciences and in 2021 held a position on the boards of two pharmaceutical companies, AmyriAD and Centessa Pharmaceuticals.
After resigning from a senior position in the FDA’s vaccine division, Philip Krause secured a role in the biotech sector. One study found more than a quarter of the FDA employees who approved cancer and haematology drugs between 2001 and 2010 left the agency and now work or consult for pharmaceutical companies.25
Beyond the FDA, Ian Hudson, chief executive of the UK’s MHRA between 2013 and 2019, now serves on the board of biotech company Sensyne Health and is a senior adviser for the Bill and Melinda Gates Foundation. Before joining the MHRA, Hudson held various senior roles at pharmaceutical giant SmithKline Beecham.
Reform
Critics argue that both small and large structural changes are necessary to help restore regulators’ ability to carry out independent decision making, free of industry influence.
Lexchin outlines several reforms for advisory committees, including that all financial COIs, including the dollar amount of payment, be disclosed along with an explanation about why these people cannot be replaced with someone without COIs. Lexchin’s suggestions align with longstanding recommendations from the US Institute of Medicine.26
Kesselheim says one crucial step is for the FDA to re-examine its approach to expedited approvals. “There needs to be more clarity about the endpoints and what the scientific basis is for choosing an endpoint.” Kesselheim says greater assurances are needed that the endpoints selected truly are “reasonably likely” to predict clinical benefit, as the FDA’s accelerated approval standard requires. For expedited drugs, “you also need to make sure that a confirmatory trial is underway at the time of approval, so that it can be completed in a timely fashion. And if it isn’t completed or the trial is negative, then you need to think about how you might pull back on the product,” he says.
Light says it is no longer possible for doctors and patients to receive unbiased, rigorous evaluations from drug regulators. He suggests setting up non-profit organisations like Germany’s Institute for Quality and Efficiency in Health Care, which was established to carry out evaluations of approved drugs that are independent of industry, rigorous, unbiased, and transparent. “The question is why weren’t drug regulators doing this trustworthy, transparent, rigorous, unbiased job in the first place?” says Light.
While historical drug disasters like sulfanilamide and thalidomide raised the stature of regulatory agencies, Light argues regulators now need their own watchdog and is calling for a drug and vaccine safety board, independent of the drug regulator, with the authority, staffing, and funds to investigate incidents of patient harm. “Countries have independent safety boards for airlines and their passengers. Why not for drugs and patients too?” says Light.